什么是Genelibs?
Genelibs是济南弘晖生物技术有限公司研发的一款在线服务的数据分析、挖掘软件, 该软件可以实现高通量数据的在线分析,高通数据库的本地化,数据在线挖掘等功能, 是一款较为新颖的在线分析系统。
平台怎么注册?
通过在线平台中的“点击注册”功能进行自助注册。点击查看/注册
平台现在主要有哪些功能?
平台主要功能就是生物数据的查询、本地化、处理和可视化展示等功能。
平台是如何收费的?
现阶段,注册用户都可以享受免费的小型模块分析服务,基本上可以满足简单的分析需求。
对于大型实验室和科研机构,我们准备了模块套装,可以因实验室定制高级模块和套装。
而针对个性化的分析和研究项目的需求,我们还有定制模块的服务,可以根据您的需要,定制开发模块,满足分析需求。
我自己没有数据,可以使用这个系统做点研究吗?
完全可以,通过数据的定制,或者数据库检索,您可以找到您所研究的疾病信息,找到疾病数据,进行分析,得到结果,指导您 的进一步实验等。
平台的分析方法靠谱吗?有参考文献没有?
所有的分析,都是现有的方法,都是公认的经典或者新颖的方法,均有参考文献,并且经过测验验证可用。
分析软件有没有bug?
软件虽然都经过严格的测试,但是仍难免出现bug,如果您在分析过程中,出现的任何您解决不了的错误和信息,请及时反馈给我们,我们会尽快修正。
什么是基因简称(symbol)
表示各基因所用的符号。基因的名称都尽量用缩写的字母来表示,并用斜体书写。如为显性基因,符号的第一个字母大写,如为隐性基因则小写(如 Pg和 pg)。标准(或野生)型的基因以 号表示(如 vg , vg 或 )。 同一座位如果有三种以上的等位基因时,必须在基本符号的肩上附记字母或数字以示区别(如 I O , I A , I B 等)。 相同表型的基因位于二个以上的座位时,应在基本符号和字母或数字间加一短横,或将字母或数字记在基本符号下面 (如 ert-a, ert-b; 这种情况,合子的基因型以分数表示,分别连锁在同源染色体上的基因分子和分母分开写(如: A b C/a B c), 属于二个以上连锁群的基因以另外的分数表示(如 A b C/a B c; DE/d e)。 M. Demerec等( 1966)提出的基因符号对细菌等微生物已广泛应用。 首先用 3个小写字母来表示遗传性状(如精氨酸生物合成基因用 arg;半乳糖发酵基因用 gal;紫外线损伤修复基因用 uvr来表示), 然后再附上一个大写字母以表示与该性状有关的各个基因(如 argA, argB; galE, galK等)。各基因的野生型均以 表示,而突变型则以号数来表示(如 arg A 、 arg A-1、 arg A-2等)。
什么是基因种类(locus group)
按照基因的作用进行的基因分类:如protein-coding gene是蛋白质编码基因,withdrawn是独立基因。phenotype是显性基因。pseudogene是伪基因。non-coding RNA是不编码RNA序列。 other是尚未定义的种类。
什么是entrez码
该基因在美国国家生物技术信息中心中的代码
什么是ensembl基因码
ensembl 是一项生物信息学研究计划,旨在开发一种能够对真核生物基因组进行自动诠释(automatic annotation)并加以维护的软件。 该计划由英国Sanger研究所Wellcome基金会及欧洲分子生物学实验室所属分部欧洲生物信息学研究所共同协作运营。
什么是refseq数据库代号
RefSeq数据库,即RefSeq参考序列数据库,美国国家生物信息技术中心(NCBI)提供的具有生物意义上的非冗余的基因和蛋白质序列。
什么是基因种类(locus_type)
locus_type,即基因所在位置的类别。withdrawn是独立基因。gene with protein product是蛋白质产物基因。phenotype only是显性基因。 pseudogene是假基因。RNA, misc是一类庞杂的小RNA分子。RNA, cluster(CRISPR RNA)是在各种细菌和古细菌(archaea)中也发现了很多成簇的、规律间隔的短回文重复序列 (clustered regularly interspaced short palindromic repeat sequences,即CRISPR序列,这就是二十多年前日本科学家发现的那个序列) 和CRISPR相关基因(CRISPR-associated genes,Cas gene),这些CRISPR序列与很多病毒或者质粒的DNA序列是互补的, 说明这套CRISPR–Cas系统很有可能是生物体抵御病毒等外来入侵者的一套特异性防御机制,就好像是另外一套适应性免疫反应系统(adaptive immune system)。 RNA, small nuclear(snRNA)是细胞内的小核RNA,它是真核生物转录后加工过程中RNA剪接体(spliceosome)的主要成分,参与mRNA前体的加工过程。 RNA, small cytoplasmic(scRNA)是真核细胞细胞浆中的小RNA,它们约有100到300个碱基,在天然状态下与蛋白质相结合,scRNA参与蛋白质的合成和运输, 如SRP颗粒就是一种由一个7SRNA和蛋白质组成的核糖核蛋白体颗粒,主要功能是识别信号肽, 并将核糖体引导到内质网。RNA, pseudogene是假基因RNA。 RNA, small nucleolar(small nucleolar RNA)是核仁小RNA(small nucleolar RNA),是近来生物学研究的热点,由内含子编码,已证明有多种功能,反义snoRNA指导rRNA核糖甲基化。 与其它RNA的处理和修饰有关,如核糖体和剪接体核小RNA、gRNA等。RNA, long non-coding(lncRNA)是长链非编码RNA,一类不表达蛋白质的基因。所占全基因组的比例与生物种间的复杂等级有着更密切的相关性. RNA, Y(Y RNA)是非编码小RNA成分RO核蛋白颗粒(RO RNP),RO RNP是由勒纳等人首次发现的,在以为系统性红斑狼疮患者的自身免疫抗体的作为目标。 region是区域基因。T cell receptor gene(TCR)是T细胞受体基因。T cell receptor pseudogene是假T细胞受体基因。RNA, transfer是转运RNA(Transfer Ribonucleic Acid,tRNA) 是具有携带并转运氨基酸功能的类小分子核糖核酸。complex locus constituent是复合基因。RNA, vault是穹窿体RNA,一种存在于穹窿体核糖核蛋白复合物中的非编码RNA,该类RNA于1986年被首次发现,除了含有8-16条短链RNA外, 还包含96个穹窿体主蛋白(major vault proteins,MVP),两个穹窿体少数蛋白(minor vault proteins,分别为VPARP和TEP1),被认为与抗药性有关。 virus integration site是病毒整合位点。endogenous retrovirus是内源性逆转录病毒。RNA, micro是一类由内源基因编码的长度约为22 个核苷酸的非编码单链RNA 分子,它们在动植物中参与转录后基因表达调控, 大多数miRNA 基因以单拷贝、多拷贝或基因簇(cluster) 的形式存在于基因组中。protocadherin是原钙粘连素一类介导细胞黏附的细胞受体。immunoglobulin pseudogene是免疫球蛋白假基因。 transposable element是转座因子,可移动因子,一类在很多后生动物中(包括线虫、昆虫和人)发现的可移动的遗传因子( 一段DNA顺序可以从原位上单独复制或断裂下来,环化后插入另一位点,并对其后的基因起调控作用,此过程称转座) 这段序列称跳跃基因或转座子,可分插入序列(Is因子),转座(Tn),转座phage。fragile site是脆性位点,是染色体上的裂隙或不连续的间断区,在此间断处,如果标本做得好的话,可以看到有一些物质穿过。 readthrough是通读基因,由于模板的突变或辅助因子的帮助,RNA聚合酶或核糖体能忽略终止信号,而继续转录或翻译。RNA, ribosomal(rRNA)是核糖体RNA,它与蛋白质结合而形成核糖体,其功能是作为mRNA的支架,使mRNA分子在其上展开,形成肽链 (肽链在内质网、高尔基体作用下盘曲折叠加工修饰成蛋白质,原核生物在细胞质内完成)的合成。immunoglobulin gene是免疫球蛋白基因。
什么是基因名称状态(status)
Approved是已经公认的,Entry Withdrawn是在绘制中的。
什么是基因染色体位置(location sortable)
基因位点在所在染色体中的位置。其中,代码中的p 指染色体短臂,q 指染色体长臂。q/p后面的数字代表区,点后数字代表带。例如9q21.3–q22是指:9号染色体长臂21区3带到22区.
什么是基因简述
关于基因的简要描述,附带的中文翻译为软件自动翻译。
什么是基因家族(gene family)
是来源于同一个祖先,由一个基因通过基因重复而产生两个或更多的拷贝而构成的一组基因,它们在结构和功能上具有明显的相似性,编码相似的蛋白质产物, 同一家族基因可以紧密排列在一起,形成一个基因簇, 但多数时候,它们是分散在同一染色体的不同位置,或者存在于不同的染色体上的,各自具有不同的表达调控模式。
什么是uniprot数据码
UniProt[1] 是 Universal Protein 的英文缩写,是信息最丰富、资源最广的蛋白质数据库。它由整合Swiss-Prot、 TrEMBL 和 PIR-PSD 三大数据库的数据而成。 他的数据主要来自于基因组测序项目完成后,后续获得的蛋白质序列。它包含了大量来自文献的蛋白质的生物功能的信息。
什么是pubmed代码
PubMed医学文献检索服务系统,其数据主要来源有:MEDLINE、OLDMEDLINE、Record in process、Record supplied by publisher等。数据类型:期刊论文、综述、以及与其它数据库链接。
什么是mgd码
mouse genome database,小鼠基因组数据库。
什么是cosmic
Catalogue of Somatic Mutations in Cancer,癌症的体细胞突变目录。
什么是omim代码
Online Mendelian Inheritance in Man,在线人类孟德尔遗传数据库。持续更新的关于人类基因和遗传紊乱的数据库。 主要着眼于可遗传的或遗传性的基因疾病,包括文本信息和相关参考信息、序列纪录、图谱和相关其他数据库。
什么是mirbase
miRBase序列数据库是一个提供包括miRNA序列数据、注释、预测基因靶标等信息的全方位数据库,是存储miRNA信息最主要的公共数据库之一。
什么是merops
MEROPS是一个关于蛋白酶(或称为肽酶)及其抑制剂的在线网络数据库。它对蛋白酶的分类方案由Rawlings & Barrett在1993年提出,对蛋白抑制剂的由Rawlingset al.在2004年提出。
什么是imgt
国际免疫遗传学信息系统,是 免疫遗传学和免疫信息学的全球基准。 IMGT是优质的知识资源。集成了专业的免疫球蛋白(Ig)或抗体, T细胞受体(TR),主要组织相容性(MH)人类和其他脊椎动物物种,并在 免疫球蛋白超家族(家族成员),MH家族(MHSF相关蛋白和免疫系统(的)RPI)脊椎动物和无脊椎动物。
什么是iuphar
IUPHAR,International Union of Pharmacology,国际药理学联合会。
什么是LncRNA数据库
长链非编码RNA数据库,Long non-coding RNA,通过多种机制发挥生物学功能,这些机制包括基因印记,染色体重塑,细胞周期调控,剪接调控,mRNA讲解和翻译调控等.
基因序列
基因序列指的是该基因中的碱基及碱基的排列顺序
基因表达
Major Tissues:主要组织 Bone Marrow:骨髓;Whole Blood:所有血液;White Blood Cell:白血细胞;Lymph Node:淋巴结;Thymus:胸腺; Brain:大脑;Cortex:皮层;Cerebellum:小脑;Retina:视网膜;Spinal Cord:脊髓;Tibial Nerve:胫神经; Heart:心脏;Artery:动脉;Smooth Muscle:平滑肌;Skeletal Muscle:骨骼肌;Small Intestine:小肠; Colon:结肠;Adipocyte:脂肪细胞;Kidney:肾脏;Liver:肝脏;Lung:肺脏;Spleen:脾脏;Stomach:胃脏; Esophagus:食道;Bladder:膀胱;Pancreas:胰腺;Thyroid:甲状腺;Salivary Gland:唾液腺;Adrenal Gland:肾上腺; Pituitary:垂体;Breast:乳腺;Skin:皮肤;Ovary:卵巢;Uterus:子宫;Placenta:胎盘;Prostate:前列腺;Testis:睾丸; Immune:免疫相关的;Nervous:神经相关的:Muscle:肌肉相关的;Internal:循环相关的;Securetory:内分泌相关的;Reproductive:生殖相关的; MicroArray:微阵列;RNAseq:转录序列;SAGE:基因表达系列分析; BioGPS: ;intensity:强度;
基因引物
Primer:引物 是一小段单链DNA或RNA,作为DNA复制的起始点,在核酸合成反应时,作为每个多核苷酸链进行延伸的出发点而起作用的多核苷酸链, 在引物的3′-OH上,核苷酸以二酯链形式进行合成,因此引物的3′-OH,必须是游离的。之所以需要引物是因为在DNA合成中DNA聚合酶 仅仅可以把新的核苷酸加到已有的DNA链上。
基因本位信息
细胞分布:显示该基因及对应蛋白质在细胞中分布的位置和数量 如CCND1是和细胞分裂有关的基因,因此其具有保守性,其主要作用区域在细胞核(有丝分裂)和细胞质(细胞膜和细胞骨架的新建)中较活跃,其异常可能引起肿瘤 RIT1是和编码转运蛋白的基因,其编码的蛋白质几乎全部在细胞膜上
GO库
gene ontology:基因本体数据库,涵盖生物学的三个方面: 细胞组分(cellular component):细胞的每个部分和细胞外环境。 分子功能(molecular function):可以描述为分子水平的活性(activity),如催化(catalytic)或结合(binding)活性。 生物过程(biological process):生物学过程系指由一个或多个分子功能有序组合而产生的系列事件。其定义有广义和狭义之分,在词义上可以区分为泛指和特指。 一般规律是,一个过程是由多个不同的步骤组成。需要指出的是,生物学过程与途径或通路(pathway)不是同一回事。
基因相互作用关系
暂时收录了5个数据库的内容
STRING:预测蛋白质间的功能相关性的一个数据库
蛋白质之间的功能联系通常可以从编码它们的基因之间的关系来推断:完成相同功能的基因组在相近的物种中表现出亲缘性,在基因组上的位置相近(在原核生物中),
并往往参与基因融合事件。STRING数据库希望依赖于全球联盟的探索和分析。因为三种类型的证据概念不同,并且预测的交互的数量是非常大的,为了能够有效的评估和比较各个预测,这是必不可少的。
因此,STRING包含基于所述不同类型的对一个共同的参考集关联,集成在每预测单个置信度得分的基准的独特的得分的框架。来推断蛋白质相互作用并用网络的图形表示提供功能性连接的一个高级视图,促进生物过程模块化的分析。
STRING不断更新,目前包含完全测序的89个基因组上的261033个同源基因。数据库预测有一半拥有至少80%的准确度。(网址http://string.embl.de/)
BioGRID:生物学相互作用数据集通用数据仓库
BioGRID是一个开放访问的面向所有主要生物物种的数据仓库,收集了许多从原始生物医学文献中精选的遗传和蛋白质相互作用文档。
在2012年9月,BioGRID包括了30多种模式生物的500,000多对人工注释的相互作用。BioGRID维护了出芽酵母、裂殖酵母和模式植物拟南芥文献的完整精选覆盖。
也支持生物医学重要性领域中的许多主题精选项目。BioGRID已经为大部分主要模式生物数据库,包括酵母基因组数据库、PomBase、WormBase、
FlyBase和拟南芥信息资源建立了相互作用和表型注释的协作和/或共享数据记录。BioGRID也积极参与到文本挖掘社区来基准测试并部署自动工具以加快精选流程。
BioGRID数据可以通过一个用户定义的交互式界面免费访问,并通过一系列广泛格式(包括PSPSI-MI2.5和制表符分割文件)批量下载。
BioGRID纪录也能够用一系列新的生物信息学工具进行研究和分析,包括一个翻译后修饰查看器,一个图形化查看器,一个REST服务和一个Cytoscape插件。
(网址http//thebiogrid.org)
IntAct:一个开源分子相互作用数据库
IntAct提供了一个开源数据库和工具包进行蛋白质相互作用的存储、描述和分析。网络界面提供了蛋白质相互作用的文本描述和图形化描述,并允许在相互作用的蛋白质的基因本体论(
Gene Ontology,GO)注释背景下浏览相互作用网络。网络服务允许直接计算访问以检索XML格式的相互作用网络。
IntAct当前包括从文献中输入并与Swiss-Prot小组协作挑选的约2200个二元相互作用和复杂相互作用,大量使用受控词汇以保证数据一致性。
(网址http://www.ebi.ac.uk/intact)。
mentha:一个从不同来源收集的完整和全面的方式的数据库
其数据来自于IMEx协会通过的手工策划的蛋白质-蛋白质相互作用数据库。汇总形成一个相互作用的包括许多生物的数据库。
mentha是一种资源,并提供了一系列工具以分析选择的蛋白质网络样的的作用关系。蛋白质相互作用数据库收录了许多蛋白质-蛋白质相互作用(PPI)发表论文信息。
当然,没有任何数据库其广度能提供了一个完整的资源。
mentha的方法是生成每个星期一致的作用(图)。最重要的是,利用所有的支持证据该程序分配给每一个相互作用的关系可靠性得分。mentha提供八相互作用组(智人,Arabidopsis thaliana,秀丽隐杆线虫,
Drosophila melanogaster,大肠杆菌K12、Mus musculus、褐家鼠、Saccharomyces cerevisiae)加上一个全球网络,足以包括所有的生物,包括那些没有提到的。
网站和图形应用程序的设计和存储使得所有mentha用户方便访问其数据。(网址http://mentha.uniroma2.it/about.php)
MINT:分子相互作用数据库
分子相互作用数据库(Molecular INTeraction database,MINT)旨在从同行评议期刊发表的工作中提取实验细节,
以一种结构化的格式存储关于分子相互作用(molecular interactions,MIs)的信息。目前MINT小组集中于蛋白质间物理相互作用的精选工作。遗传的或计算推断的相互作用不包含在数据库中。
在过去的4年里,MINT已经经历了大量修正。新版本的MINT基于一个完全改造的数据库结构,它提供了更高效的数据探索和分析,并以具有一个更丰富的注释条目为特征。在过去的几年里,
精选的物理相互作用数目已经飙升至超过95,000。整个数据集能够通过基于网络的界面以交互式和批量模式在线访问和一个FTP服务器访问。作为一个整合的添加,MINT现在包括HomoMINT,
在模式生物中具有直系同源蛋白质(从实验中推断的人类蛋白质间)的相互作用的一个数据库(网址http://mint.bio.uniroma2.it/mint/)。
Reactome:一个生物通路知识库
Reactome是一个精选的、同行评议的人类生物过程资源。考虑到一个生物体的遗传组成,可能反应的完整集合构成了它的反应组。
Reactome数据库的基本单位是反应;反应然后被分组成因果链以形成通路。Reactome数据模型允许我们表征人类系统中的许多不同过程,包括中间代谢通路,调控通路和信号转导,
及高层次过程,例如细胞周期。Reactome提供了一个定性框架,定量数据能够在该框架上被叠加。Reactome开发了工具以促进专业生物学家进行定制数据录入和注释,并允许完结的数据集作为一个交互式过程图进行可视化和探索。
虽然我们的主要精选领域是来自人类的通路,但是我们通过推断的直系同源定期地创建人类通路的电子映射到其他生物体,因此使得Reactome与模式生物研究社区相关。数据库在遵循开源条款下可以公开使用,
这允许它的内容和它的软件基础设施被免费使用和重新分发。(网址http://www.reactome.org)
什么是microRNA
MicroRNA (miRNA) 是一类内生的、长度约为20-24个核苷酸的小RNA,其在细胞内具有多种重要的调节作用。每个miRNA可以有多个靶基因,而几个miRNA也可以调节同一个基因。 这种复杂的调节网络既可以通过一个miRNA来调控多个基因的表达,也可以通过几个miRNA的组合来精细调控某个基因的表达。据推测,miRNA调节着人类三分之一的基因。最近的研究表明大约70 %的哺乳动物miRNA 基因是位于TUs区 ( transcription micro RNA micro RNA units , TUs ) ( Rodriguez et al ,2004) , 且其中大部分是位于内含子区( Kim &Nam , 2006) 。 一些内含子miRNA 基因的位置在不同的物种中是高度保守的。miRNA 不仅在基因位置上保守, 序列上也呈现出高度的同源性(Pasquinelli etal , 2000 ; Ruvkun et al , 2001 ; Lee & Ambros ,2001) 。 miRNA 高度的保守性与其功能的重要性有着密切的关系。miRNA 与其靶基因的进化有着密切的联系, 研究其进化历史有助于进一步了解其作用机制和功能。
什么是基因通路图
基因通路:人体基因组图谱好比是一张能说明构成每一个人体细胞脱氧核糖核酸(DNA)的30亿个碱基对精确排列的“地图”。科学家们认为,通过对每一个基因的测定,人们将能够找到新的方法来治疗和预防许多疾病,如癌症和心脏病等。 该图非常形象地把基因家族的各种基因描绘出来。 20世纪50年代以后,随着分子遗传学的发展,尤其是沃森和克里克提出双螺旋结构以后,人们才真正认识了基因的本质,即基因是具有遗传效应的DNA片断。 研究结果还表明,每条染色体只含有一个DNA分子,每个DNA分子上有多个基因,每个基因有含有成百上千个脱氧核苷酸。由于不同基因的脱氧核苷酸的排列顺序(碱基序列)不同,因此,不同的基因就含有不同的遗传信息。 KEGG pathway:(Kyoto Encyclopedia of Genes and Genomes)KEGG(京都基因与基因组百科全书)是基因组破译方面的数据库。在后基因时代一个重大挑战是如何使细胞和有机体在计算机上完整的表达和演绎, 让计算机利用基因信息对更高层次和更复杂细胞活动和生物体行为作出计算推测。为达到此目的,人们建立了一个在相关知识基础上的网络推测计算工具。在给出染色体中一套完整的基因的情况下, 它可以对蛋白质交互(互动)网络在各种细胞活动起的作用作出预测。 KEGG 的PATHWAY 数据库整合当前在分子互动网络(比如通道,联合体)的知识,KEGG 的GENES/SSDB/KO 数据库提供关于在基因组计划中发现的基因和蛋白质的相关知识, KEGG 的COMPOUND/GLYCAN/REACTION数据库提供生化复合物及反应方面的知识。 Reactomepathway:反应组学(Reactome, http://www.reactome.org)是一个汇集了由专家撰写,经同行评阅的有关人体内各项反应及生物学路径的文章的数据库,该数据库相当于一个有效的数据资源以及电子图书。 该库目前发布了共计2975个人类蛋白、2907项生物学反应以及4455个引用文献。该数据库为人们提供了一个全新的从整体水平上对生物学途径进行研究的工具,同时,它也是一个改良的搜索及数据挖掘工具,可以简化与生物学途径相关的数据搜索与研究。 此外,对用户提供的高通量数据组进行分析,也变得更为简单。目前,由于直系同源预测方法的改进,反应组学数据库也开始收录其它模式生物的数据了,现在通过与其它数据库合作和人工注释方式,已经收录了包括拟南芥(Arabidopsis)、 水稻(Oryza sativa)、果蝇(Drosophila)及原鸡(Gallus gallus)等22种模式物种的反应组学数据。反应组学的数据库内容和相关软件都是开源共享,免费使用的。
什么是基因相关疾病
因相关基因异常可能引起的疾病或导致疾病的发生概率提高.Befree数据库:Bio-Entity Finder and Relation Extraction。 BeFree利用文本挖掘技术工具来解锁包含在生物医学的文档的信息。 BeFree是一个畅通的生物医学命名实体识别(BioNER)的模块(基于使用fuzzy-模式匹配方法来查找和唯一标识实体在文献中提到的字典),它也被用于基于支持向量机(SVM)的关系抽取(RE)。 BeFree被用于从科学出版物的生物提取的关联信息的文本挖掘工作流程。简言之,文档选择之后,文本挖掘方法包括两步。第一步是生物医学出版物的实体的识别和归入BioNER模块的装置,第二步,由RE模块进行处理以识别上述的实体之间的关系。
什么是基因相关文献
genelibs收录的相关文献均来自已发表刊物。
什么是基因评论
基因评论系统是开放式的评论系统
平台使用时数据本地化失败
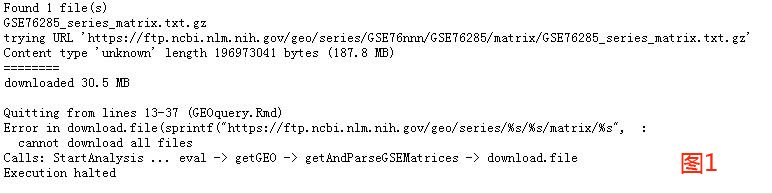
问题:网络原因,数据未下载全.
处理:重新运行
平台使用时取子集错误
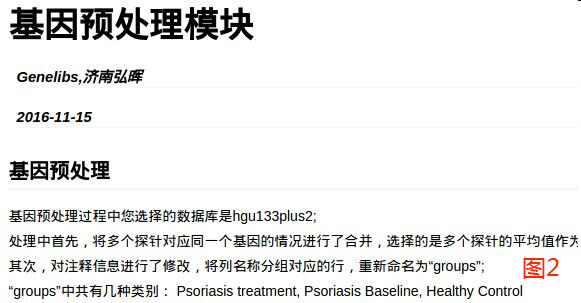
问题:分组信息中名称不一致(大小写及空格)
处理:取子集过程中输入的组名要与表达集数据信息处理过程中更改后的名称保持一致。
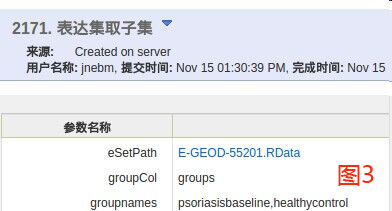
平台使用时分组信息中名字或样本编号有空格,不能直接使用
处理:利用“预处理”模块进行转换
步骤:
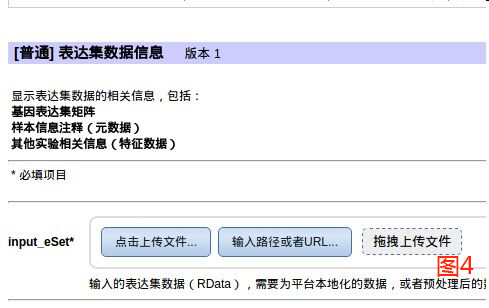
1、将RDATA放入“表达集数据信息”(图4),点击运行生成两个csv文件(图5)

2、打开pDatamatrix.csv,找到其中的分组信息所在的列,更改列名称(groups)及组别名称信息(如去除空格等),并保存
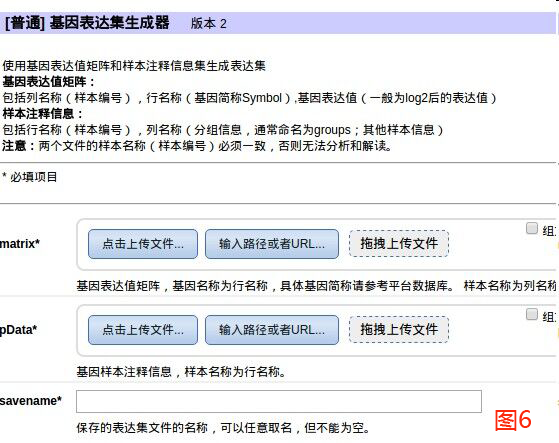
3、将修改好的pData文件上传到“基因表达集生成器”中(图6),点击运行生成新的RData(图7)
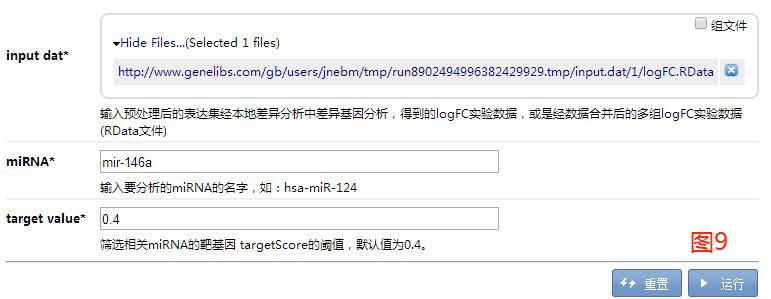
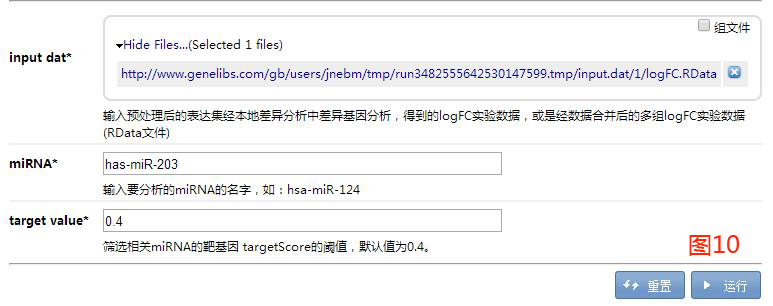
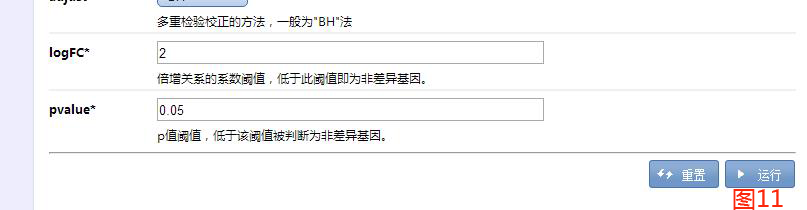
常见的miRNA相关靶基因预测时犯的错误